���ӄ����Wģ�M�Ǹ߷��ӌW�����о���Ч�Pϵ����Ҫ�ֶΣ���㕽ӌ�����Փ����Ҫ������Ȼ�������yȫԭ�ӷ��ӄ����Wģ�M���g��������ģ�M�Ŀ��g���r�g�߶��������ޣ����y�M�㌍�H��������ģ�M���g������Ч�ʵ���Ҫ;���������ڴ������^�������ɶȵļs����ȱ����C-H�I�ȵ��^���W��������Ч���]�����@�õĶ�߶ȽY���������W�����|�c����ȫԭ��ģ�M���ƫ���^��ֻ�������붨��Ӌ���c�A�y���O��������˷��ӄ����Wģ�M�ڽ�ጌ���¬F�������²����аl�ȷ���ĝ��ܡ���ˣ�ؽ��lչ���W�خ��Ծ��ʴ�����ģ�M�������ڼs���wϵ���ɶȵ�ͬ�r����Ч���]C-H�I�ȱ��s�����ɶȌ��wϵ������|��Ӱ푣����F�߷����wϵ��ͬ�r�ճ߶ȽY���������W���|�Ĝʴ_�����������@Ҳ��Ŀǰ�߷�����Փģ�M�I�����R���������y�}����Ҫ���Hǰ�ط���
�D1 ���ڏV�x��֮�f���̵ĸ߷��Ӵ�����ģ��

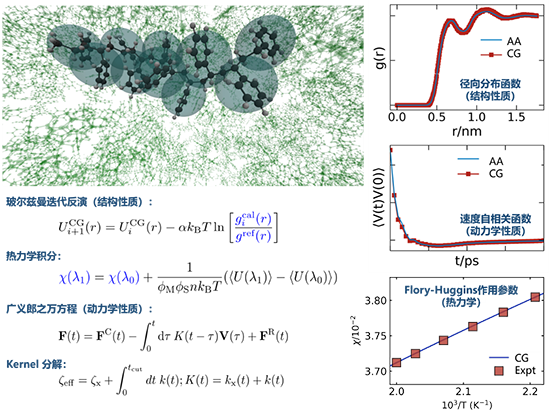
���ڣ����ִ�W�X��܊���ڈF������܉�ʴ_���������g����ֲ������ĵ�������Ɲ�����ݷ����c�V�x��֮�f������Y�ϣ��������뱻�s������C-H�I�Ȼ��W�������wϵ�����W���|��Ӱ푣����ú��rӛ���Ⱥ˺�������ȫԭ��ģ�M�Ы@�õ��ٶ������P������~�����O������˴�����ģ�����Ծ���λ�ơ��ٶ������P�������������P�����������ɳ�ģ���Ȟ�����Ą����W���|���@�ýY���cȫԭ��ģ�Mһ�¡����˼���ʶ��cӋ��Ч�ʣ�ԓ���������s���L�r�gӛ���Ⱥ˲���һ���R���ɷ��RouseĦ��ϵ�����Լ����R���ɷ�Ķ̕r�gӛ���Ⱥˣ�ǰ�������L�r�g�ĔUɢ�О飬���ߛQ���˾ֲ�����ײ�c���Uɢ�О顣ͬ�r�������_�l��һ��ϵ�y���ă������̣����ڕr�g��ه��ӛ���Ⱥ˵Ę�����ԓ�����������ƏV������ď��s�ۺ����wϵ���ڴ�����ģ�������_���Ľ��^ģ�M�߶ȣ�ᘌ��ض��wϵ�����F����ȫԭ��ģ�M���ȵĻ��W�خ��ԵĶ�߶ȽY���������W���|���A�y��
ȫ��朽ӣ�https://doi.org/10.1021/jacsau.3c00756
