趙宏宇 研之成理 2025年05月13日 10:09 浙江
全文速覽
本研究基于密度泛函理論(DFT)計算預測,通過采用溫度調控的熔鹽輔助合成策略,成功構建了由正交相和立方相RuTe2(m-RuTe2)組成的異相同質結催化劑。理論研究表明,該異相同質結構通過電荷平衡效應、優化的Ru-Te軌道雜化程度及氫吸附能。在堿性條件下,m-RuTe2在10 mA cm-2電流密度下僅需35 mV的析氫反應過電位,這一性能不僅顯著優于單相RuTe2催化劑,更超越了商業Pt催化劑的活性。此外,得益于Ru-Te結構單元之間強烈的電子耦合作用,該催化劑有效抑制了Ru活性中心的溶解,表現出優異的長期穩定性。
![]()
![]()
背景介紹
氫氣因其高能量密度和綠色環保特性,被視為極具發展潛力的可持續能源。在催化領域,與傳統異質結構催化劑相比,異相同質結構催化劑展現出顯著優勢:其單一的化學組成確保了活性中心的均勻分布,從而顯著提升了催化活性、穩定性和選擇性。更重要的是,異相同質結構中兩相間的功函數(WF)差異可誘導形成內置電場,這一特性不僅促進了電子轉移和電荷再分布過程,還能優化活性中間體的吸附行為,最終實現催化性能的顯著提升。
![]()
![]()
本文亮點
1、RuTe2異相同質結兩相之間的功函數差誘導了內建電場的形成。
2、內置電場驅動的電荷轉移和重排優化了RuTe2在反應中間體上的吸附。
3、采用溫度調節熔鹽輔助策略制備了異相同結結構RuTe2催化劑。
4、RuTe2異相同質結在堿性介質中表現出優異的析氫反應活性和長期穩定性。
![]()
![]()
圖文解析
圖1. (a)c- RuTe2和o- RuTe2的功函數。(b)功函數誘導的電子轉移示意圖。(c)m- RuTe2的差分電荷密度分布和Bader電荷分析。(d)m- RuTe2的功函數。(e)RuTe2中Ru、Te的分波態密度(PDOS)。(f)m- RuTe2中的電荷轉移過程。(g)堿性HER的吸附自由能圖
DFT理論計算結果揭示,通過構建正交相(o-RuTe2)與立方相(c-RuTe2)的異相同質結模型(m-RuTe2),可顯著提升析氫反應(HER)活性。兩相間顯著的功函數差異誘導產生強的內置電場,該電場不僅驅動界面電子的定向轉移和重新分布,還優化了反應位點的電子結構,從而促進了氫中間體的吸附-解離平衡。相較于單一晶相,m-RuTe2的異質界面在降低水解能壘的同時還優化了m-RuTe2表面的氫吸附自由能ΔGH*。這表明催化劑電子結構的優化可顯著加速析氫反應動力學(圖1)。
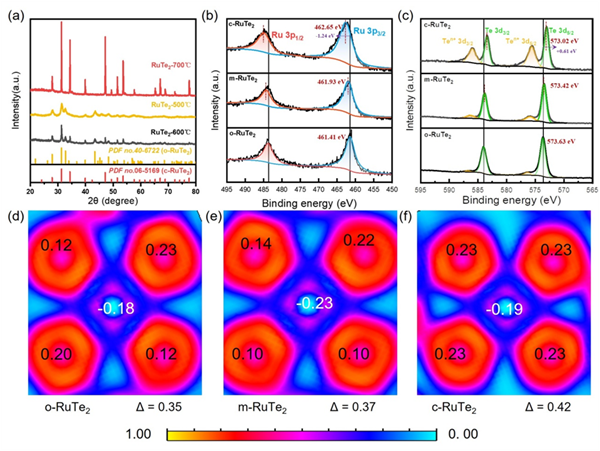
圖2.(a)所有合成材料的XRD圖譜。(b)所有合成材料的Ru 3p的XPS光譜。(c)所有合成材料的Te 3d的XPS光譜。分別從(d)o-RuTe2、(e)m-RuTe2和(f)c-RuTe2觀察價電子的電子局域函數(ELF)的2D切片,數值是Ru和Te的計算Bader電荷值。
通過溫度調控的熔鹽輔助法,成功實現了不同RuTe2晶相的可控合成。XRD分析表明,500 ℃條件下合成的樣品呈現正交相結構(o-RuTe2),700 ℃的產物為立方相(c-RuTe2);而在600 ℃這個中間溫度下,樣品顯示出o-RuTe2和c-RuTe2兩相共存的特征(m-RuTe2)。值得注意的是,XPS分析顯示,隨著合成溫度從500 ℃升高至700 ℃,Ru與Te之間的電荷轉移程度逐漸增強,同時Ru的氧化態也隨之升高。進一步研究發現,與單一晶相的o-RuTe2或c-RuTe2相比,兩相共存的m-RuTe2展現出獨特的電子結構特征。這主要源于兩相間功函數差異誘導產生的內建電場促進了界面電荷的轉移與重排,從而形成了適中的Ru-Te軌道雜化程度。這種優化的電子結構為催化反應提供了有利條件。電子局域函數(ELF)分析進一步證實,從o-RuTe2到c-RuTe2存在逐步增強的電荷轉移趨勢,與實驗結果一致。(圖2)
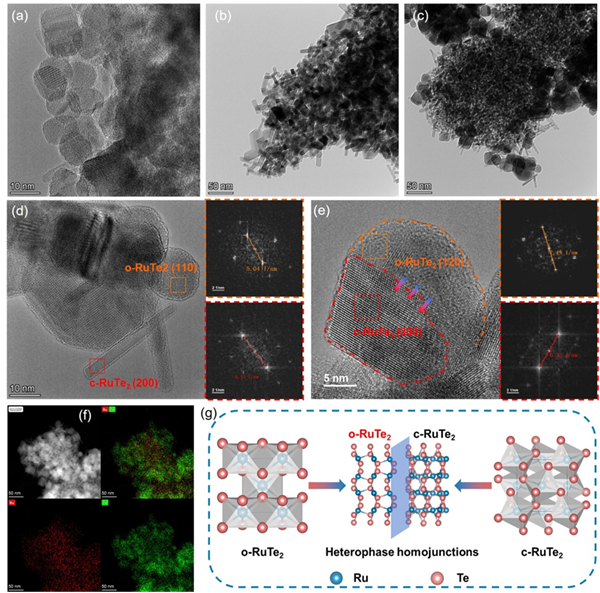
圖3. (a)o-RuTe2、(b)c-RuTe2和(c)m-RuTe2的TEM圖像。(d-e)m-RuTe2的HRTEM圖像和相應區域的FFT圖案:橙色:o-RuTe2,紅色:c-RuTe2。(f)m-RuTe2的EDS作圖。(g)異相同質結結構形成示意圖。
TEM圖像分析表明,三種RuTe2樣品呈現出顯著不同的結構特征:o-RuTe2以球形納米顆粒為主,c-RuTe2表現為典型的棒狀納米結構,而m-RuTe2則展現出獨特的"球-棒"復合結構,即大量細小的棒狀納米顆粒生長于球形納米顆粒表面。HRTEM圖像進一步揭示了m-RuTe2中兩相共存的結構特征,并觀察到了從c-RuTe2向o-RuTe2的相變過渡區域。這一結果直接證實了RuTe2異相同質結構的成功構建。(圖3)

圖4.(a)所有不同RuTe2樣品的LSV曲線(掃描速率:10 mV s-1)。Pt/C用作參比。(b)每個樣品的Tafel斜率和(c)奈奎斯特圖。(d)各種催化劑的Cdl。(e)各種催化劑在50 mV過電位下的質量活性和比活性。(f)m-RuTe2的恒定電壓穩定性測試。(g)m-RuTe2//RuO2和Pt/C//RuO2朝向整體水分解。(h)m-RuTe2//RuO2的恒定電壓穩定性測試。
采用標準三電極體系在1.0 M KOH電解液中評估了不同RuTe2樣品的析氫反應性能。其中,單斜相m-RuTe2表現出最優異的催化活性,僅需35.3 mV的過電位即可達到10 mA cm?2的電流密度,顯著優于正交相o-RuTe2(62.3 mV)和立方相c-RuTe2(164.3 mV)。此外,m-RuTe2的反應動力學和電化學活性表面積(ECSA)均優于o-RuTe2和c-RuTe2,并展現出比商業Pt/C更優的質量活性和比活性。m-RuTe2在20小時的恒電流電解過程中,電流密度保持率達91%;經過2000次循環伏安(CV)加速老化測試后,其過電位僅增加2 mV,展現出出色的長期穩定性。在m-RuTe2||RuO2雙電極體系中,在2.28 V的電壓下即可驅動1000 mA cm?2的電流密度,優于商業Pt/C||RuO2體系性能(2.36 V)。同時,該體系還還表現出優良的長期穩定性。(圖4)

圖5.(a)HER測試后所有合成材料的XRD圖譜。(b)測試前后m-RuTe2的Ru 3p的XPS光譜。(c)不同RuTe2催化劑連續催化反應后溶解在電解質中的Ru和Te的濃度。(d)不同RuTe2催化劑中Ru的脫金屬能量。(e)不同RuTe2催化劑中Ru-Te能帶的-COHP(f)不同RuTe2催化劑中Ru-Te能帶的積分-COHP至費米能級比較。(g)形成抑制反應性金屬溶解的異相同質結結構的兩相的示意圖。
通過對比HER測試前后的XRD圖譜,發現m-RuTe2的晶體結構保持完好,特征衍射峰無明顯變化;XPS分析進一步證實其表面化學態保持穩定。ICP-OES定量分析顯示,在20小時恒電位電解后,m-RuTe2中Ru和Te的溶解濃度低于o-RuTe2和c-RuTe2。這些測試結果表明,m-RuTe2異相同質結構具有增強的結構穩定性。DFT計算進一步揭示了該穩定性增強的機理:首先,m-RuTe2中Ru的溶解能壘高達9.54 eV,高于o-RuTe2和c-RuTe2;其次,晶體軌道哈密頓布居(COHP)分析表明,m-RuTe2具有最強的Ru-Te共價相互作用(-ICOHP = 2.79 eV)。這種增強的穩定性源于異相界面處內置電場驅動的電荷再分配與強化的Ru-Te共價耦合的協同效應:內置電場促進界面電子離域,而增強的共價作用則提供了結構骨架支撐,二者共同抑制了金屬溶解,有效緩解了長時間HER過程中的結構退化問題(圖5)。
![]()
![]()
總結與展望
本研究成功設計并構建了由立方相和正交相組成的異相同質結(m-RuTe2)催化劑。兩相共存誘導的內置電場效應顯著促進了電荷轉移與重排過程,進而優化了活性中間體在催化劑表面的吸附行為,從而加速了催化反應的動力學進程。此外,Ru-Te之間的強電子耦合作用顯著抑制了析氫反應過程中Ru活性位點的失活和溶解問題。實驗結果顯示,m-RuTe2異相同質結催化劑具有遠優于單相RuTe2和商業Pt催化劑的催化活性和長期穩定性。這一重要發現不僅證實了異相同質結結構設計在提升傳統單相催化劑性能方面的獨特優勢,還極大地拓展了催化劑的研究維度,為其他催化體系的材料設計提供了新的研究思路。