���������ڮ�������������ռ������Ҫ�ĵ�λ���䑪�÷������w�˿��������ߡ��r�ñ�Ĥ���t����е���I��ȫ�������������^��7000�f����Ŀǰ������Ʒ���Ķ�Ԫ����Ͷ�Ԫ���M�����ڿs�Ͼۺ��Ǵ�Ҏģ���I���a���������;�����������I���ձ���ý��ٴ������������Q������Ó���^���Ķ�Ԫ������߮a���������Ȼ�����@Щ�����^�����ﶾ�ԵĽ��ٴ����������ھ������w�У������w����Ȼ�h�����Σ��������1929�꣬�߷��ӌW�Ƶ����Wallace H. Carothers���о��˶�Ԫ�����c��Ԫ������������w�Դ����M�����������@����������������Ҫ����κδ�����Ȼ���a��������H��2~5 kDa������̫����o�����á���һ�����o���ڴ������������h����Ӱ�Խ��Խ�ܵ���ҕ�ı����£������о��Դ������s���y�ԫ@�ø߷������������挍ԭ��̽������Դ��s��Ч�ʵ��C������;�����ص�Q�����д��������Ć��}�������ش���Փ���x�͑��Ãrֵ����Ҳ���R������
�����W���أ������������ĵ�ƽ�ⳣ�������w�����ų����a��ˮ�����y�����ձ��J���nj����Դ������o���@�ø߷�����������ԭ��Ȼ�������I�ϏV��ʹ�õ������Q�����ڽ��ٴ������«@�ø߷������ľ����a����������Q������ƽ�ⳣ��(< 1)��������������(~4)��С�����a��(���Ҷ���)��ˮ���y�ų����@���f���˟����W���ؑ��������Դ����o���@�ø߷������a���ԭ���ڄ����W�ϣ��@�ø߷���������߀Ҫ���_��������F��Ħ�����@�ӵĿ��̗l�������Դ����������������������������c�u����ȵ�һ�η����Ȼ���ȵĶ��η������ȡ����H�����^���У����H���ڻ��F����½����º��ھۺ������^�������Ҵ�����F����ƫ�x��Ħ���ȣ����¾����ķ���期o�����L���@Ҳ���ǂ��y�����Q������Q����Ҫ���}������������W���ؑ����������Դ��¾����a���������ߵ��P�I��

�������������J�R���㽭��W�߷��ӿƌW�c���̌Wϵ��ε豸������F������һ�N���͵ğo�����s��(CFP)�C����ԓ�C��������һ��܉��γ���Ԫ�h������Ԫ�h�����Ķ�Ԫ����������w(�D1)��
�^���Ĵ��Ԫ���c����Ԫ�������γ��Ȼ�����A����l�����µĴ�������
-
(1)�Ȼ�����A����l������ķ��ӃȺͷ����g�|���D�ƣ��քe�γ��|�ӻ����Ȼ���x�Ӻ��Ȼ������x�ӣ�
-
(2)�����ĩ�˵��Ȼ������x�ӡ���ҧ�����ɭh���������������u�����F��
-
(3)�������u�����Ȼ���x���ٴ��M������������Ó�����a��ˮ��ʹ�÷���朰l�����L��
ͨ�^������������ʹ���wϵ�еĴ�����F�Ȳ���څ����1:1���a����������L�ʬF�����صġ����١�ģʽ���Ķ����c���y��ˇ����ĕr�g�ȣ�ͨ�^���ڿs�۫@����һϵ�еĸ߷������o���������������۶����ᶡ������(PBS)���۶������Ҷ�����(PES)����(�����ᶡ������-��-�����ᶡ������) (PBSA)�;�(�������Ҷ�����-��-�����������Ҷ�����) (PEST)�����̘I���Ŀɽ���������ɴˣ��C���˂��y�J֪�ğ����W���y�����nj����Դ����о����������y����ߵ���Ҫԭ��Ŀǰԓ�о��ɹ����}��Catalyst-free synthesis of polyesters via conventional melt polycondensation���о�Փ�İl����Materials Today�ϣ������x������Փ����
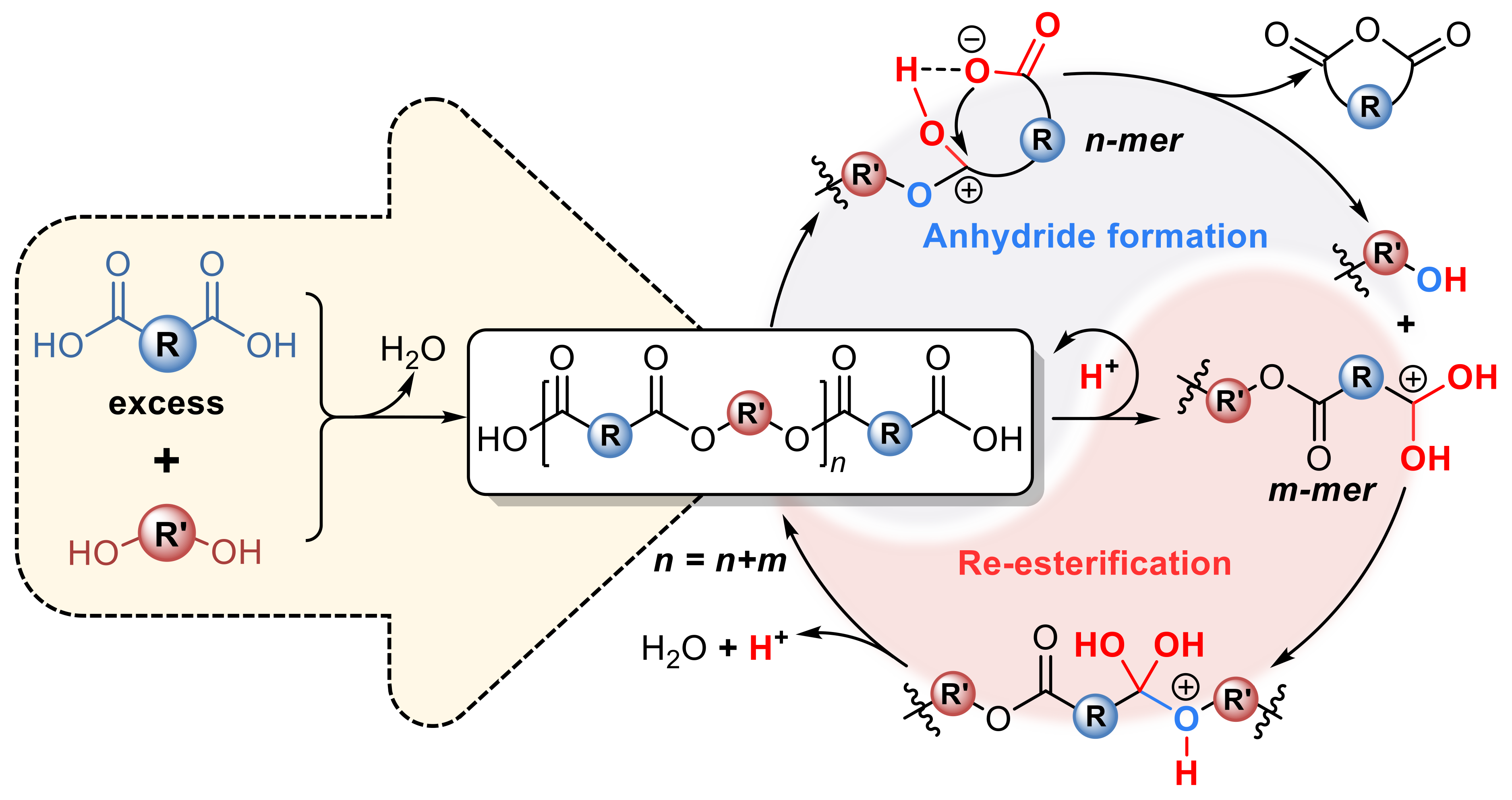
�D1. CFP���ϳɾ����ęC��
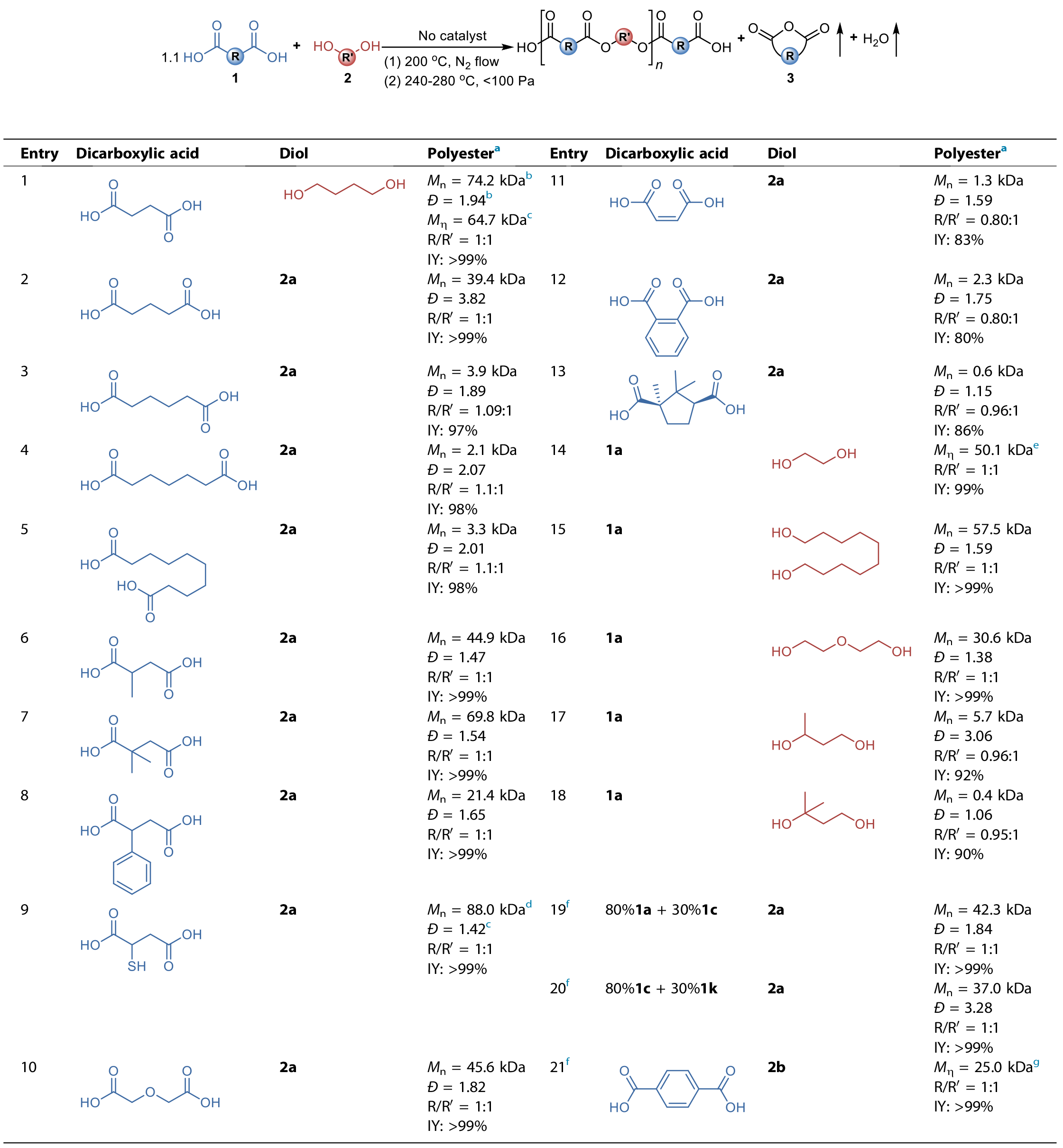
�������Ȍ������C���M�����о���ʹ��Ħ�����^��10%�Ķ�����(1a)�c1,4-������(2a)�ڟo��Ӵ�����200��C�ėl���M�������������@�����Ȼ���˵�PBS�A���1H NMR�V�D(�D2a)�C����PBS�A�����еĶ������Ԫ�c��������Ԫ֮��(ӛ��R/R��)��1.11:1���f����Ĵ_���Ȼ���ˡ�ԓPBS�A������Ĕ���������(Mn)����������(M��)�քe��1.2��1.3 kDa���S���˴_�J�Ƿ����ͨ�^PBS�A�����ĩ�˵ġ���ҧ���a�������������A�����������]��ƿ�в���240��C�������¼ӟ��������_��ƽ�⡣�Y���l�F���ڼӟ��Ļ�����1H NMR�D�V��(�D2a)�����س��F�˴�����������(3a)�ķ壬���������ڶ�Ԫ����Ԫ���京��(6%)�cʣ��Ķ������Ԫ����(105%)֮�̈́����c�ӟ�ǰ�Ķ������Ԫ����(111%)�Ǻϡ��c���γɌ��ȵ��ǣ��ڼӟ�ǰ��PBS�A�����Ў��z�y�������������Ĵ��ڡ�ԓ�Y��ʹ����K�a��ķ������@��������3�����_����Mn = 3.6 kDa��M�� = 3.5 kDa�����ң�PBS�A�����MALDI-TOF-MS�V�D������ϵ�еķ����x�ӷ嘋��(�D2b)���������弰��������С��w���ڃɶ˾����Ȼ��ľ����(P1)���c�x�Ӻ���x�Ӽӳ��������ϵ�е�С����һ�˞��Ȼ�һ�˞��u���ľ����(P2)���c�x�Ӽӳ�����߰l�F��P2�ij��m�Ԯa���Ǿ����������܉����L���P�I�����C������ͬ�ȵķ����l���£�ʹ�ö������cĦ�����^��10%��1,4-�������M�з����r�����wϵ�в�δ̽�y��P2�Ĵ��ڡ����ң����y���γ������ļ�����(1c)���c���wϵ���^�쵽�������Ҫ��P1�͞�������P2��ֻ�ИO�����������Y���C������朶����ɭh�����������Ǿ����������ܷ����L���P�I��

�D2. �����C���о���(a)PBS�A����(��)������240��C���]�ӟ�������ƽ���(��)��1H NMR�V�D����; (b) MALDI-TOF-MS�V�D�@ʾPBS�A�����Ƀɶ˾����Ȼ��ķ����(P1)�cһ���Ȼ�һ���u���ķ����(P2)����
��������������������ķ����̶ȣ����ߌ����l��������ߜغ����(240 ��C, < 100 Pa)����ԓ�l���£�PBS�A�����еĴ����Ԫ��(R/R��)��5 h�ȏij�ʼ��1.1:1�����ӽ�1:1����K�a������������_����64.7 kDa (��1��entry 1)���@Ҳ�f����ͨ�^��Ԫ������Դ����@�ø߷�������������ȫ���еġ�Ȼ�������ۼ����ᶡ������(PBA)�A���ﰴ��ͬ�ӵėl�������l�F�a��Ĵ����Ԫ��R/R����δ�a�����@׃������K�Ĕ���������Ҳֻ��3.9 kDa (��1, entry 3)�������Mһ������N��Ԫ����Ͷ�Ԫ�����w���m�����M������C�����磬�����γɭh�����������(1b)��(��)-��������(1f)��2,2-����������(1g)��(��)-����������(1h)��(��)-����������(1i)�Ͷ��ʴ���(1j)�c1,4-�����������ɫ@�ø߷������a�� (��1, entry 2��6~10)�������L̼朵��y�γ������Ķ�Ԫ�����������(1d)�����(1e)����K�a����������A�ڵ��^��(��1, entry 4��5)��Ȼ���l�F�����N��������Σ��������R����(1k)������������(1l)��(+)-���X��(1m)���й�ܗ�����p�h�Y���Ķ�朶�Ԫ��r���s�H�ܫ@�õ;��� (��1, entries 11~13)�������@Щ�Y�����������a��ķ�������ԓ�c�����γɷ��������������������������P����������ڃɶ˶��Dz��u���Ķ�Ԫ�����ɫ@�ø߷������a��(��1, entries 14~16)����ʹ�þ������u���������u���Ķ�Ԫ��������Ҳ�H�ܫ@�õ;���(��1, entries 17��18)��
��1. ���ö�N��Ԫ���c��Ԫ�����w�ϳɟo��������
����߀ͨ�^�ܶȷ�����Փ(DFT���D3a)Ӌ����������γɷ�������Փ������(����G?)����Փ�������(����G?max)�����քe�����y���ķ���ƽ�ⳣ��K�ͷ������ʳ���k1�Č������D���Y���l�F����֮�g���и߶ȵľ������P��(�D3b��3c)���܉�@�ø߷������������wϵ�У����m�˵Ħ���G?�ͦ���G?max�^�g�քe��4.61~10.97 kcal·mol-1��45.74~55.29 kcal·mol-1�������ܫ@�ø߷��������wϵ���t���ڴ˅^�g�⡣�ɴˣ�ͨ�^Ӌ��������Փֵ�Ƿ����ں��m�^�g�����܉��A�y������͵Ķ�Ԫ������m���ԡ������ԭ�����������γɷ����������܉��c������������������ƥ�䣬�����^���^�͵������γ����ʾ��o���@�ø߷������a����������J�R������߀���o�����s�۷��ƏV���y���γɻ�o���γ������Ķ�Ԫ������w��ͨ�^�c���γ������Ķ�Ԫ����w�Ĺ��s�ۣ�ʹ�������ɲ����������������܉�ƥ�䣬�ɹ��ث@���˸߷�������PBSA����(�����ᶡ������-��-�R���ᶡ������)��PEST(��1��entries 19~21)��

�D3. DFT�о������A�y�ԡ�(a)DFTӋ��������γ��^�̵�������׃��; (b)��ՓӋ��Ħ���G?�c���y����lnK�ʸ߶Ⱦ������P��(c)��ՓӋ��Ħ���G?max�c���y����lnk1�ʸ߶Ⱦ������P
�������Ҳ�u�������ϳɵğo�������ܷ��_���̘I�aƷ�Ę˜ʡ��D4a��չʾ�˺ϳɵğo����PBS�c���N�̘I��̖��PBS������˹��H1200������GS pla AZ91TN���Ѻ�Bionolle 1001MD��������(M��)�͔���������(Mn)���ȡ��Y���@ʾ���o����PBS���@��헅������^�˰�˹����������ƷPBS�����Ѻ͵�PBSʹ�þ������ﶾ�ԵĶ����������M�ДU朣��mȻ�_������ߵ�M�� = 116.0 kDa���������ܵ����ơ��D4b�@ʾ���o����PBS���@4�N��Ʒ��չʾ����ߵ����쏊��(49.22 �� 5.61 MPa)���^�õĔ������L��(32.63 �� 5.87%)������߀���o����PBS�wϵ�ʹ��^���������QPBS�wϵ�ľۺτ����W�M�Ќ���(�D4b)�����y�Ĵ��^���������Q�wϵ�����u����������(-d[OH]/dt)�c�u�����([OH])��ѭ�����Pϵ���ɴˮa��ķ����������ڷ����r�g��Mt = kt+M0�����o���������wϵ�У��u����������(-d[COOH]/dt)�c�Ȼ����([COOH])��һ�N��ϼ����Pϵ���a��ķ������c�����r�g���Pϵ�飺Mt = C(ekt-1)+M0 (C��k�鳣��)��������������L�ʬF�����صġ����١�ģʽ���mȻ����ʼ�r�o���������ķ��������L���ʵ���Sb2O3��SnCl2���ăɂ��wϵ��Ȼ���ھۺϺ���(> 4.5 h)�o���������ķ������������^��SnCl2�����wϵ����o�����wϵ�Юa��������Ҳ���Ԍ�����ղ����c����һ���ۺ�ѭ�h��(��D4d)����ˣ�ͨ�^�@�ӵ�һ�Nѭ�h�Ϳ��Ԍ��Fԭ�ӵĸ�Ч���ã����ҿɱ����ˮ����ĸ��a��Įa����ʮ�ַ��ϡ��Gɫ���W����ԭ�t�����⣬����С�� MC3T3-E1 ǰ�ɹǼ����� L929 ���w�S�������o����������PBS��PES���۹���ᶡ������(PDS)��PBSA��PEST �;�����ᶡ������(PBG)�M�еļ�������ԇ�会������ʷքe�� 94.2-102.4% �� 93.7-99.3% �ķ����ȣ��C���˟o�����������������������ԡ�

�D4. PBS�ı��^�c�������ա�(a)�o����PBS�c���N��Ʒ������𤔵([��])�ͷ���������; (b)�o����PBS�c���N��ƷPBS���������܌���; (c)�o����PBS�c���y���^���Ľ��ٴ���(Sb2O3��SnCl2)����PBS�ľۺτ����W����; (d)�����ğo�������ڿs�ۺ������Ļ���ѭ�hʾ��D
�C�ϣ������ڌ���Ԫ�����Դ��o���@�ø߷�����������ԭ�����J�R���������ǂ��y�J֪�ğ����W����(���������ĵ�ƽ�ⳣ�������wϵ��Ó�����a��ˮ�����y)���µģ��������ڴ����Ԫ���ڄ����W��ƫ�x1:1������ģ������һ�N�ϳɸ߷������o����������CFP�C�������в�����һ����Ԫ�����^���Ŀ��γɭh�����Ķ�Ԫ����������w���ڂ��y�ăɲ����ڿs�۹�ˇ�£����ȫ@��һ�N�Ȼ���˵��A����S��ͨ�^�������Ļ�Ԫ�������|���D�ơ������γɺ��ٴ������M�з����������L��ԓ�C��Ҳͨ�^������Ӌ��C��ՓӋ��@�ó���C�������ң��@�NCFP������չ���o���γ������Ķ�Ԫ����w���猦�������ᡣͨ�^�c���γ������Ķ�Ԫ��Ĺ��s�ۣ��Ϳ��ԫ@��һϵ�еĸ߷�������������ԓ�������H�܉����a�c�F���̘I�aƷ�ஔ�ğo�������������ҏصױ����˴���������ؓ��Ч����ʮ�����������ھ��и߰�ȫ��Ҫ����I��(���t����е���������)�������ڲ����ӳɱ�����r�¸��®�ǰ�ľ�����ˇ��
ԓՓ�ĵĵ�һ���ߺ�ͨӍ���߷քe���㽭��W�߷���ϵ2021�ò�ʿ���I������Ȫ(�F���ڻ��W�c���������V�|ʡ����ң��������о��T)����ε豸��������㽭��W�߷���ϵ������ڡ���ʿ���I����������ʿ������ܣ��Լ��㽭��W���όWԺҦ��ϼ��ʿ��ԓՓ�ĵĹ�ͬ���ߡ�ԓ�о��ܵ��ˇ�����Ȼ�ƌW������㽭ʡ�ܳ����������Y����
Փ����Ϣ��Qiuquan Cai, Tianwen Bai, Hongjie Zhang, Xuxia Yao, Jun Ling, and Weipu Zhu*. Catalyst-free synthesis of polyesters via conventional melt polycondensation. Mater. Today, 2021.
https://doi.org/10.1016/j.mattod.2021.07.024
- ����|�����пƴ���Ƙ��� Adv. Sci.���ߺ����ۺ��һ偷����B�m����-���F�����ɻ���̻������ߺ����;����Ч�ϳ� 2025-05-30
- ��ƴ����F� Angew���Y�� / �����Ӧ�-����������ƽ���_�h�ۺ��Ƃ���Ѓ���������ܵ��]��ѭ�h���������� 2025-05-14
- ���ڴ�W���ɷ������悵� Macromolecules: �p�Y�c���w��會Aб�����Ӱ푳��߷���������ϩ���S�����^���еľ��wȡ�� 2025-04-14
- ����Ȫ�n�}�M Adv. Mater.���U�f������������������²��Լ���100L�������ȳɹ����� 2025-06-08
- ��˹�˴�WAlejando M��ller/����������ġ������M Biomacromolecules���ɽ���������ۣ�������-co-ʮ����������Ĺ��Y���О� 2025-05-21
- ��ƴ����c�����O�����F� Macromolecules��ˮ��Passerini���M�ַ�ɢ�ۺϷ����Ƃ������ 2025-05-20
- �㽭��W��ε豽��� Adv. Mater.���U�fPET�o�����o�܄����������Ƃ�����オ������ 2024-09-03
